Homoseksüellik veya diğer adıyla eşcinsellik aynı cinsiyete duyulan cinsel, romantik veya erotik duygu ve davranış biçimidir. Homoseksüellik sadece insanlarda değil doğada neredeyse her canlı türünde görülen bir cinsel davranış biçimidir.
Homoseksüellik biyolojik bir gerçektir
Aynı cinse veya karşı cinse olan seksüel yönelim psikolojik veya çevre kaynaklı sosyal bir olgu olmadığı bilakis biyolojik temeli olan bir gerçeklik olduğu artık biliniyor.
– Peki, cinsel yönelimi yani heteroseksüel, biseksüel veya homoseksüel olmayı belirleyen şey nedir?
Bu konuda yapılan araştırmalar homoseksüelliğin genetik, epigenetik, doğum öncesi rahim içi hormonların etkisi ve nörolojik kaynaklı olduğunu gösteriyor.
Aşağıda okuyacağınız bu makale iki bölümden oluşuyor. Birinci bölümde cinsiyet nedir, kaç çeşit cinsiyet vardır, cinsel yönelim nedir, kaç çeşit cinsel yönelim vardır gibi toplumda pek bilinmeyen, bilinse de pek konuşulmak istenmeyen konulara yer verilecek ve ardından geçmiş yıllarda homoseksüellik konusunda yapılmış genetik çalışmalardan bazı örneklere başlıklar halinde değinilecek. İkinci bölümde ise Chicago Üniversitesi tarafından yapılan ve 7 Aralık 2017 tarihinde Nature dergisinde yayınlanan çok önemli bir araştırmanın dikkat çeken ayrıntıları anlatılacak.
1. Bölüm
İkiden fazla cinsiyet var
Toplumda insanların kadın ve erkek olmak üzere iki cinsiyetten oluştuğu konusunda neredeyse değişmez bir inanışı bulunmaktadır. Halbuki yapılan genetik çalışmalar bu basit anatomik sınıflandırmanın dışında gri alanda kalan ikiden çok daha fazla cinsiyetin olduğunu gösteriyor. Bunlar her ne kadar genetik mutasyonlar sonucunda oluşan nadir sendromlar olsa da bunları görmezden gelemeyiz, yok sayamayız.
Üçüncü gruptaki cinsiyetlere birkaç örnek
- Genetik olarak erkek, görüntü olarak kadın olan Swyer Sendromlular (XY-Kadını),
- 47 XXY, 46 XY /47 XXY kromozom setine sahip olan Klinefelter Sendromulular,
- 45,X0 veya 45,X/46,XX kromozom setine sahip olan Turner-Sendromlar, üçüncü grupta bulunan cinsiyetlerden sadece birkaçını oluşturmaktadır.
Gerek kromozom, gerekse gen düzeyinde meydana gelen değişikliklerden oluşan bu tür cinsel gelişme bozuklukları (disorders of sexual development) istatistiksel olarak 4500 doğumda bir görülür.
Son yıllarda yapılan çalışmalarla gri alanda kalan cinsiyet tanımlamalarına yenileri de eklenmekte. Örneğin, her yüz doğumda bir görülen hypospadias yani üretranın penisin ucunda ve altında bulunması göre yapılan ince tanımlamaların da cinsel gelişim bozukluklarına dahil edilmesi ile cinsiyet çeşitliliğindeki sayı daha da artıyor. Özetleyecek olursak; Kadın ve erkek cinsiyeti dışında gerek genetik gerekse anatomik farklardan kaynaklanan birçok ara cinsiyet çeşidi bulunmaktadır. (1)
Not: Bazı batı ülkelerinde parlamento düzeyinde 3. grup cinsiyetler için toplumsal bazı düzenlemeler yapılıyor.
Kaç çeşit cinsel yönelim vardır
Yapılan araştırmalar cinsiyet çeşitliliğinde olduğu gibi cinsel yönelimde de geniş bir yelpazenin olduğunu gösteriyor. (2)(3)
İndiana Üniversitesi‘nden biyolog, seksolog, entomoloji ve zooloji profesörü Alfred Charles Kinsey’in 1948 ve 1953 yılları arasında yapmış olduğu araştırma ile homoseksüellik ve heteroseksüellik arasında 5 farklı cinsel yönelim grubunun daha tanımladı. Alfred Charles Kinsey’in kendi adıyla anılan Kinsey skalasındaki (The Kinsey Scale) cinsel yönelim grupları şöyle:
- Tamamen heteroseksüel
- Baskın heteroseksüel, nadiren homoseksüel
- Baskın heteroseksüel, sıklıkla homoseksüel
- Eşit derecede heteroseksüel ve homoseksüel
- Baskın homoseksüel, sıklıkla heteroseksüel
- Baskın homoseksüel, nadiren heteroseksüel
- Sadece homoseksüel
- Cinsel temas yok

Homoseksüellik geni aranıyor
Yapılan birçok çalışma her ne kadar homoseksüelliğin genetik kaynaklı olabileceğinin ipuçlarını verse de bu konuda etkili kaç genin olduğu ve bu genler üzerinde kaç varyantın olduğu halâ net olarak bilinmiyor.
Esas konuya geçmeden önce hazır yeri gelmişken homoseksüellik konusunda geçmiş yıllarda yapılan önemli araştırmalardan birkaçını başlıklar halinde verelim.
- Karşılaştırmalı beyin taramalarında gerek geylerin gerekse lezbiyenlerin beyin yapısının heteroseksüel akranlarından farklı olduğu tespit edildi. (4)
- İkizlerle ilgili yapılan birçok çalışma cinsel yönelimin genlerle bağlantılı olduğunu işaret ediyor. (5)
- Homoseksüel erkek kardeşler ile yapılan çalışmalar geylerin % 67’sinin X kromozomunun Xq28 bölgesinde farklılıklar olduğunu gösteriyor. (6)
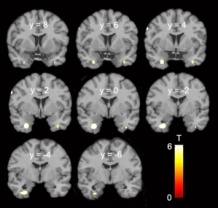
- Genom çapında yapılan diğer araştırmalar homoseksüel erkeklerde sadece X kromozomunun Xq28 bölgesinin farklı olmadığını, bunun dışında 7. kromozomun 7q36, 8. kromozomun 8p12 ve 10. kromozomun 10p26 bölgelerinin de farklı olduğunu gösteriyor. (7)
- Kore Bilim ve Teknoloji Enstitüsü tarafından dişi fareler ile yapılan ilginç bir başka araştırma ise cinsel yönelimde genlerin ne kadar etkili olduğunu gösteren bir başka örnek. Bu araştırmada FucM geni tamamıyla iptal edilen dişi farelerin diğer dişi farelere karşı erkeksi cinsel davranış sergiledikleri belirlendi. (8)
- X kromozomu ile yapılan bir başka çalışmada erkeklerde homoseksüellik ile ilgili genlerin anne kaynaklı olduğunu işaret ediyor. Buna göre homoseksüel erkekler anne tarafında, baba tarafına göre daha çok homoseksüel kuzen ve dayıya sahipler (Bu oran anne tarafından %13 dayı, baba tarafından %6 amca). (9)
- Erkek kardeşlerin doğum sırasının da erkeklerde cinselliği etkileyen epigenetik bir başka faktör olduğunu gösteriyor. Fraternal birth order effect denilen bu durumda, her doğan erkek çocuk kendisinden sonra doğacak erkek kardeşinin homoseksüel olma ihtimalini yaklaşık % 33 oranında artırıyor. (10)
- Prenatal androjen model olarak adlandırılan bir başka epigenetik faktör ise hamilelik süresince anne tarafından salgılanan veya anne tarafından ağız yolu ile alınan androjen hormonunun doğacak olan kız çocukların gelecekte lezbiyen olma ihtimalini artırdığını gösteriyor. (11)

2. Bölüm
Dikkat çeken iki yeni mutasyon keşfedildi.
Genetik varyasyonların, yani SNPs lerin geniş çaplı araştırıldığı Genome-wide association study adlı proje kapsamında yapılan çalışmada, 1077 homoseksüel ve 1231 heteroseksüel erkeğin DNA örnekleri incelendi ve deneklerin genomlarında meydan gelen tek harflik mutasyonlar SNPs karşılaştırıldı. Yapılan gen analizleri sonunda genomun iki farklı lokusunda (bölgesinde) erkeklerde cinsel yönelimi etkileyebilecek anlamlı iki gen varyantı yani noktasal mutasyon SNPs keşfedildi ve bu iki noktasal mutasyonun homoseksüel erkeklerde heteroseksüellere göre daha yaygın olduğu tespit edildi.
Cinsel yönelimin karmaşık genetik yapısı göz önüne alındığında bu iki gen varyasyonunun homoseksüelliğin anlaşılmasında iyi bir ipucu olacağı söyleniyor.
Nedir bu mutasyonlar
1. Mutasyon (Lokasyon: Chr13: 86504577 – 86504577, rs9547443 SNV C -> T)
Birinci mutasyon 13. kromozomda SLITRK5 ve SLITRK6 genleri arasında yer alıyor. Bunun ne anlama geldiğini ancak SLITRK geninin görevinin ne olduğunu anlayarak anlayabiliriz.
SLITRK geninin görevi nedir?: SLITRK Gen Ailesi, nöronal gelişimde önemli rol oynayan bir gen grubudur. Önceki yıllarda yapılan araştırmalarda bu gen ailesinin nöropsikiyatrik davranışlarda da önemli rol oynadığı tespit edilmişti.
SLITRK geninin nöropsikiyatrik davranışlarda rol almasından yola çıkan araştırmacılar bu genin cinsel yönelimde de etkili olabileceği düşünerek SLITRK genini mercek altına aldılar ve genin özellikle hipotalamus ve talamus arasında yer alan Diensefalon’da (ara beyinde) aktif olarak çalıştığını buldular. Daha sonra yapılan beyin taramasında beynin bu bölgesindeki hücre yoğunluklarının homoseksüel ve heteroseksüel erkeklerde farklı olduğu tespit edildi.
Sonuç olarak SLITRK6 genindeki tek harflik bir mutasyon protein yapısının değişmesine ve buna bağlı olarak da ara beyindeki hücre yoğunluğunun farklılaşmasına sebep oluyor. Bu farklılığın cinsel yönelimi nasıl etkilediği konusu henüz tam olarak bilinmiyor. Bunu için de ayrı bir araştırma yapılması gerekiyor.
2. Mutasyon (Lokasyon: Chr14: 81445087 – 81445155, rs4411444 SNV A -> G)
Homseksüellik ile Guatr/Tiroid bağlantısı: Keşfedilen bu ikinci mutasyon, homoseksüel erkeklerin 14. kromozomunda tiroid hormonu sentezleyen TSHR reseptör geninde bulunuyor. Bu gende meydana gelen değişiklikler tiroid bezinin aşırı çalışmasını ve buna bağlı olarak da Basedow hastalığı (Toksik guatr) gibi tiroid hastalıkların ortaya çıkmasına sebep oluyor. Yapılan bu çalışmada TSHR geninin, hipokampüs dahil olmak üzere 10. kromozomun çeşitli bölgelerinde de aktif olarak çalıştığı tespit edildi (12).
İlginç olan başka bir konu ise daha önce yapılan çalışmalardaki bulgulardı. Bu bulgular bazı tiroid rahatsızlıkları ile homoseksüellik arasında bir bağlantı olabileceğini gösteriyordu. Örneğin, Danimarkalı araştırmacıların bu konuda yaptığı bir araştırma homoseksüel erkeklerin heterosexuel erkeklere göre daha fazla tiroid rahatsızlıklarına yakalandıklarını gösteriyordu ve ayrıca hamilelik süresince tiroid rahatsızlığı çeken annelerin daha fazla homoseksüel erkek çocuk doğurdukları elde edilen bulgular arasındaydı. ( 13)
13)
Konu hâlâ çok spekülatif
Genome-wide association study çerçevesinden yapılan bu çalışmadan elde edilen bulgular halâ çok spekülatif. Çünkü araştırmaya katılan denek sayısı çok az. Her ne kadar denek sayısı az olsa da elde edilen bulgular bize homoseksüelliğin muhtemel genetik bağlantıları konusunda bazı ipuçları veriyor. Bu muhtemel bağlantıları güçlendirmek için daha çok sayıda katılımcının genetik bilgilerine ihtiyaç var. Bu amaçla Genome-wide association study çerçevesinde daha geniş çaplı yeni araştırmaların hazırlıkları başladı.
Sonuç
Homoseksüellik, evrimin milyonlarca çeşitliliğinden sadece birisidir. Ne özenti ile ne de yetiştirilme ile homoseksüel olunur. Araştırmalardan edinilen bulgular bize homoseksüelliğin bir tercih değil aksine genlerimizin dikte ettiği bir yönelim olduğunu söylüyor. Bilim bize aynı zamanda “Homoseksüel olarak doğulur, başka bir ifade ile sonradan homoseksüel olma diye bir şey yoktur“ diyor.
Tarih boyunca birçok toplumda homoseksüellere karşı neredeyse faşizme varan ayrımcılık uygulandı ve halâ az gelişmiş ve tutucu toplumlarda bu insanlık dışı uygulama devam ediyor. Bilim, teknoloji ve tıp alanındaki gelişmeler her konuda olduğu gibi homoseksüelliğin biyolojik sebeplerini anlamamıza ışık tutuyor ve bu bağlamda her geçen gün toplumların homoseksüelliğe bakış açısı yavaş yavaş değişiyor. Artık özellikle batılı toplumlarda homoseksüelliğin bir hastalık veya ahlaki bir sorun olmadığı, aksine biyolojik bir gerçek olduğu biliniyor.
Bu konuda son olarak şunu söylemek istiyorum: Homoseksüellik binlerce yıldır vardır ve canlılık var olduğu sürece de var olacaktır. En muhafazakar bir ailede de, en çağdaş bir ailede de homoseksüel bir çocuk doğabilir. Buna biz değil genlerimiz karar verir.
Benzer konuda hazırlanmış diğer makaleler
- Kromozomal bozukluklar ve cinsiyet belirsizliği
- Genetik el falı / Parmak uzunluğu ne anlama geliyor ?
- Eşcinsellik doğuştan mı?..
- Erkekte parmak uzunluğu ile hastalıklar ve penis uzunluğu arasındaki ilişki.
Mehmet Saltuerk
++++++++++++++++++++++++
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics
of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
- Sex redefined
- Bisexualitäten – Geschichte und Dimensioneneines modernen wissenschaftlichen Problems
- Interview mit dem Soziologen Rüdiger Lautmann: “Nicht nur Sex”
- Homosexual Women Have Less Grey Matter in Perirhinal Cortex than Heterosexual Women
- Homosexual behavior due to genetics and environmental factors
- Linkage between sexual orientation and chromosome Xq28 in males but not in females
- A genomewide scan of male sexual orientation
- Male-like sexual behavior of female mouse lacking fucose mutarotase
- A linkage between DNA markers on the X chromosome and male sexual orientation
- H-Y Antigen and Homosexuality in Men
- Finger-length ratios and sexual orientation
- A Link Between Maternal Thyroid Hormone and Sexual Orientation?
- Genome-Wide Association Study of Male Sexual Orientation
Bu blogdaki makaleler bir başka yayın organında kaynak gösterilmeden yayınlanamaz, çoğaltılamaz ve kullanılamaz.



































 balığının ürettiği Skualamin ise Alfa-Synuclein’in yanlış katlanmasından kaynaklanan bütün bu olumsuzlukların önüne geçiyor
balığının ürettiği Skualamin ise Alfa-Synuclein’in yanlış katlanmasından kaynaklanan bütün bu olumsuzlukların önüne geçiyor
{kind=link}