Sağlıklı bir hücrede, hücre bölünmesi katı kurallara göre gerçekleşir. Her yeni bölünmede kromozomlar yeni hücrelerin içerisine ikişer adet olacak şekilde dağılırlar. Bu sağlıklı bir dağılımdır. Ancak kanserli hücrelerde ve Down sendromu gibi bazı genetik hastalıklarda bu katı kurala tam olarak uyulmaz. Bu kromozomal istikrarsızlık kanser hücrelerinin tipik özelliğidir.
Anöploidi hücreler
Hücre dejenerasyonu genellikle hücreler içindeki kromozomların yapısal ve sayısal durumunda farklılıklar şeklinde kendini gösterir.
Kanserli hücreler bölünme esnasında bazen bir veya iki kromozom kaybeder bazen de sayılarını istedikleri gibi artırırlar. Bu nedenle kanser hücreleri ilerleyen zamanda farklı kromozomal değişiklikler gösterirler ki, bu durum hem tümörü daha agresif hale getirir hem de tedaviye karşı daha dirençli yapar. İşte vücutta kromozom dengesinin bozulmasına sebep olan ve anormal sayıda kromozom içeren bu tür hücrelere Anöploidi hücreler denir.
Aslında Anöploidilerin kanserdeki yaygınlığı yüzyılı aşkın süredir bilinmesine rağmen, anöploidilerin tümör gelişimindeki rolü tartışmalı olmaya devam etmektedir. Hücre içerisinde normalden fazla bulunan kromozomlar, kansere sebep olan genlerin dozajını arttırarak tümörü teşvik ettiği ileri sürülmüştür ama bu tezin kanıtları oldukça eksiktir.
Bu konuda bir başka tez ise ilerlemiş kötü huylu tümörlerde (malignite) sıklıkla meydana gelen kontrol noktası kaybının (loss checkpoint control) bir sonucu olarak anöploidi lerin ortaya çıkabileceği öne sürülmüştür. Gerçekten de, 21. kromozomun üç kopyası bulunan Down sendromlularda ağır kansere yakalanma riski önemli ölçüde azdır. Bu da bazı durumlarda anöploidinin tümör baskılayıcı özelliklere sahip olabileceğini düşündürmektedir.
Dev kanser hücreleri (polyploid giant cancer cells)

Bir çok Anöploid hücre bir araya gelerek Anöploidi dev kanser hücrelerini oluştururlar. Bu hücrelerin kromozom sayıları normal tümör hücrelerinin kromozom sayılarından 4, 8 hatta 16 kat daha fazla olabilmektedir. Dev kanser hücreleri genellikle standart kemoterapi veya radyasyon tedavisine dirençlidirler. Bu nedenle kanserin metastaz yapmasında belirleyici bir katkı sağlarlar. Tedaviye dirençli üçlü negatif meme kanseri, bazı prostat kanseri türleri ile bazı yumurtalık kanseri türleri dev kanser hücrelerinden oluşurlar.
Poliploid dev hücrelerin hücre yapılarında özellikle çok sayıda aktin lif bulunur ve bu lifler kanserli hücrelerin hareket etmelerine yardımcı olurlar. Dev kanser hücreleri bu lifler sayesinde daha yavaş ama daha uzağa göç etme kabiliyetine sahiptirler. Bu da metastazın daha uzak bölgelerde görülmesine sebep olur.
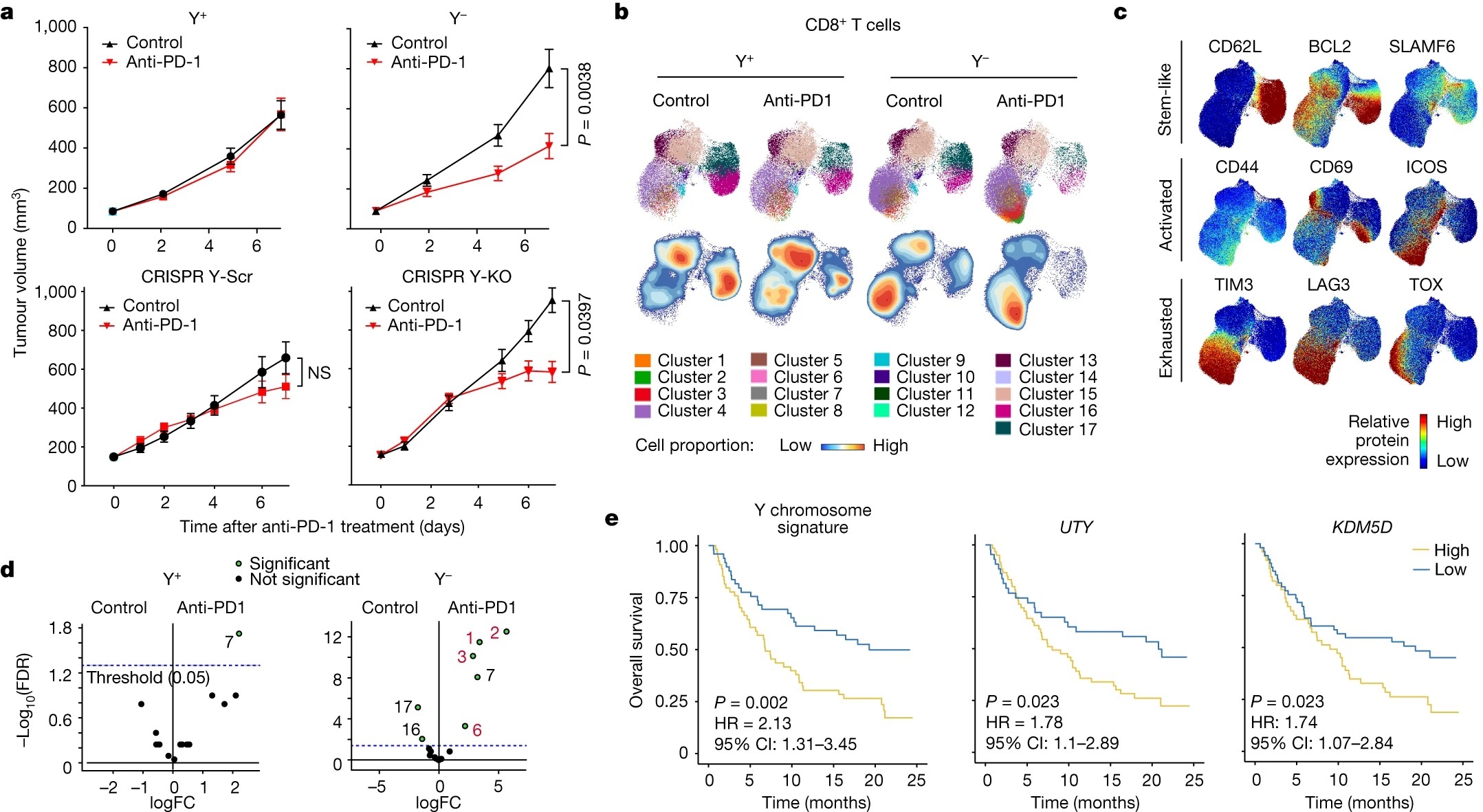
Ekstra kromozomlar kanserli tümörleri neden daha agresif hale getiriyor ?
Bu sorunun yanıtı 23.500’den fazla kanser hastasının verileri incelenerek yapıldı. Yapılan araştırmanın sonuçları, kromozomların veya onlara ait bazı parçaların extra fazladan kopyasının bulunduğu tümör hücrelerinin diğer normal kanser tümörlerinden daha hızlı büyüdüğünü gösterdi.
Açıklama: Bir tümör hücresi içerisinde, 1. kromozomun q kolu da dahil olmak üzere bazı DNA bölümleri normalden daha fazla kopya halinde bulunuyorsa, bu, birçok kanser türünde tümör büyümesini destekler. Bu veri, çeşitli kanser türlerinin hücre kültürleri ile yapılan testlerde doğrulandı. Yapılan testlerde CRISPR tekniği kullanılarak 1. kromozomun q kolunun fazladan üçüncü kopyası tümörlü hücrelerden çıkarılarak tümörlü hücrenin kromozom sayısı normalleştirildi.
Sonuç: Normalleştirilmiş kopya sayısına sahip tümörlü hücreler daha küçük tümörler üretti, hatta bazen hiç yeni tümör oluşturmadı.
Not: Bu araştırmada, meme kanseri gibi bazı agresif tümörlerinde gelişen 1. kromozom q kolu anormallikleri ele alınmıştır. Diğer kanser türlerinde gelişen başka kromozom anormallikler araştırılmamıştır ancak diğer kanser türlerinde de mekanizmanın aynı olduğu tahmin edilmektedir.
Extra kromozom artışının sebebi bazen kanserli hücrenin bizzat kendisi de olabiliyor
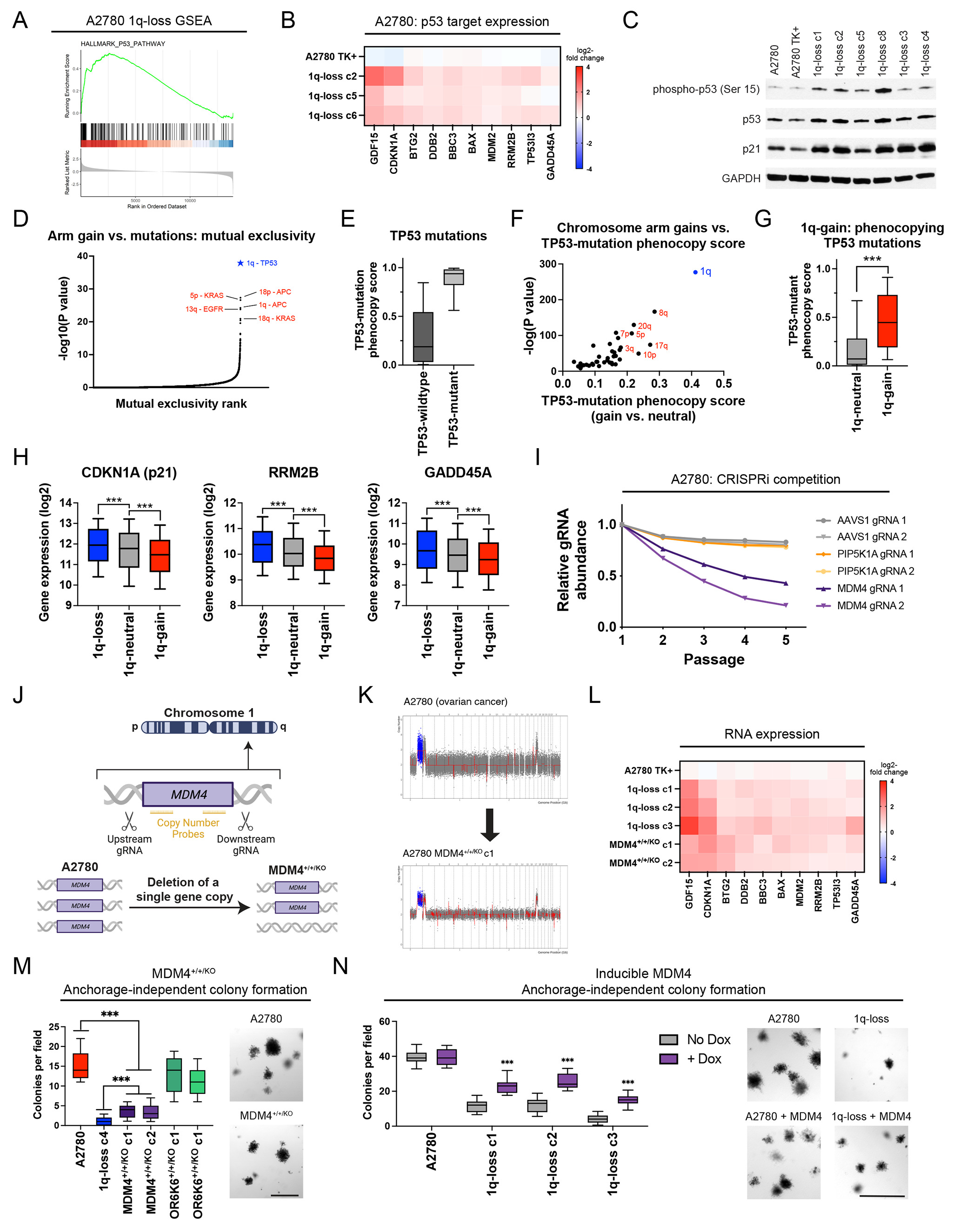
İki kromozoma sahip normal meme kanseri hücreleri kimi zaman kanserin ilerleyen safhalarında 1. kromozomun q kolunu ekstra kopyalayabiliyorlar. Bu da bize Anöploidi’in yani çoklu kromozomun sadece kanser büyümesi için gerekli olmadığını, aksine büyüyen tümörlerin de bizzat kedisinin bu oluşumu teşvik ettiğini gösteriyor. Başka bir ifade ile söylemek gerekirse; Eğer bir kanser tümörü 1. kromozomun q kolunun 3. kopyasına sahipse bunu mümkün olduğunca korumaya özen göstermektedir. Bu durum hücre kültürü deneylerin ile de sabitlenmiştir.
Extra kromozom artışı kanseri neden teşvik ediyor
Bu soruya gen ekspresyonu analizleriyle cevap arandı. Bunun için 1. kromozomdaki onkogenlerin faaliyetleri incelendi.
MDM4 gibi kanseri teşvik eden bazı genler, kanser hücrelerinde üç kopya halinde 1. kromozomun q32.1 kolunda yer almaktadır. Kanserli hücrelerde bulunan extra fazladan kromozomlar her hücre bölünmesinde sayılarını arttırır iken aynı zamanda üzerinde bulunan bu onkogenlerin sayısını da arttırdılar. Gen ekspresyonu analizleri, onkogenlerin sayısının artması ile gen ekspresyonu sonucu ortaya çıkan proteinlerin de arttığını gösterdi. Sonuç olarak artan onkogen proteinlerine paralel olarak tümörlerin hem daha hızlı, hem de daha büyük olduğu tespit edildi.
Onkogenlerin artan aktivitesi aynı zamanda 17. kromozomda bulunan TP53 geninin sentezledigi ve tümör baskılayıcı görevi olan p53 proteinin de çalışmasını engelledi. Bu da vücudun kanserle mücadelesini zayıflatan bir faktördür.
Özetleyecek olursak; Kanserli hücrelerde yer alan fazladan her kromozom (anöploidi), aynı zamanda fazladan onkogenleri de bulundurmaktadır. Onkogenlerinin sayı olarak fazla olması tümörlerin çok çabuk ve hacim olarak çok daha büyük olmasına sebep olmaktadır ki, bu da kanserin şiddetini artıran bir faktördür. Araştırmacılar, bu mekanizmanın diğer onkogen bulunduran kromozomlarda da devreye girerek tümör büyümesini teşvik edebileceğinden şüpheleniyorlar.

Risk ve fırsat aynı anda
Yukarıda belirtildiği gibi birçok kanser hücresinde gereğinden fazla kromozomun olması kanseri daha agresif hale getirmektedir. Ancak Science dergisinde yayınlanan bu çalışmanın sonuçları gereğinden fazla kromozom kopyalarının kanserle savaşta yeni bir umut olabileceğini de gösteriyor. Çünkü her fazlada kromozom kopyası sadece kanseri teşvik eden genlerin değil aynı zamanda kansere karşı koruyucu genlerin sayısını ve onların aktivitesini de artırmaktadır. Bu da kanser hücrelerini, tedavide kullanılan aktif maddelere karşı daha duyarlı hale getirebilmektedir. Örneğin artan gen aktivitesi daha fazla membran pompası, protein veya enzim üretilirse, bu kemoterapide kullanılan ilaçların alımını kolaylaştırarak etkisini artırabilir. Bu durum, 1q anöploid kanserli hücreler ile yapılan araştırmada doğrulandı. Araştırmada kanser önleyici bir enzim olan UCK2‘nin miktarı kanserli hücrelerde çok fazla miktarda üretildiği görüldü. Ayrıca yapılan araştırmada iki kemoterapötik ajanın daha miktarının artığı görüldü.
Bu araştırma anöploidinin kanser terapileri için potansiyel bir hedef sunulabileceğini gösteriyor. Eğer extra fazla kromozoma sahip kanserli hücrelere özel, hedef odaklı kemoterapik ajanlar geliştirilir ise sağlıklı dokulara zarar vermeden kanser ile mücadele daha etkin bir şekilde yapılabilecek. Burada belirleyici olan, kanserli hücre içerisinde fazldan kopyalanan kromozomlarda bu oluşumları teşvik eden genlerin bulunup bulunmaması. Zira her kromozomda farklı genler bulunmaktadır.
Benzer konularda hazırlanmış diğer makaleler
- Y kromozomu kaybı erkeklerde kanseri daha agresif yapıyor
- Beyin kanseri
- Bazı kanser türlerinde erkeklerdeki X Kromozomu devre dışı bırakılıyor
- Kanser vakalarında metastaz oluşumunu engelleyen madde: ANGPLT4
- Asparagin diyeti meme kanserinin yayılmasını durdurabilir
- Şeker, meme kanseri için büyük risk oluşturuyor.
- B6 ve B12 vitaminleri akciğer kanseri riskini yükseltiyor
- Boy ile diyabet 2, kalp damar hastalıkları ve kansere riski arasındaki ilişki
- Düşük dozda Resveratrol bağırsak kanserine karşı daha etkili.
- Sabun yapımında kullanılan Triclosan karaciğer kanserine yol açabilir !
- Özel bir sprey ile birkaç dakikada kanser teşhisi
- Kahve, rahim içi kanser riskini düşürüyor.
- Tüm kanser vakalarının % 40 ı önlenebilir.
- Aspirinin bağırsak kanserine karşı koruyucu etkisi
- Otizme karşı kanser ilacı?
Mehmet Saltuerk
++++++++++++++++++++++++
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics
of the University of Cologne
++++++++++++++++++++++++
Kaynak
Bu blogdaki makaleler bir başka yayın organında kaynak gösterilmeden yayınlanamaz, çoğaltılamaz ve kullanılamaz.