Tanım
Ehlers-Danlos Sendromları (EDS), bağ dokusunu etkileyen nadir görülen heterojen bir hastalık grubudur*. Hastalığa kolajen sentezinin bozulması neden olur. Kolajen sentezinin bozulması ise bağ dokusunun bozulmasına sebep olur.
Bağ dokusundaki kusurlar ciltten kemiklere, eklemlerden kan damarlarına kadar birçok organ ve dokuda yaşamı tehdit eden komplikasyonlar ve semptomlara neden olur. (Heterojen bir hastalık grubu*. Aynı özelliğin (fenotip) farklı genler tarafından ortaya çıkma olgusudur.)
Epidemiyoloji
Ehlers-Danlos sendromu her iki cinste de aşağı yukarı eşit oranda görülür. Hastalığın toplumda görülme sıklığının 1: 5.000 ile 1: 10.000 arasında olduğu tahmin edilmektedir.
Semptomlar
Kollajen sentezinin bozulmasına bağlı olarak bağ dokusu bakımından zengin olan deri, kan damarları ve eklemler yetersiz gelişir ve bu da güç eksikliği, bağ dokusunun aşırı gerilmesi ve özellikle kan damarlarının kolay yırtılmasına neden olabilir. Bağırsak yırtılmaları, fıtık, omurga eğriliği ve tekrarlayan pnömotorakslar* mümkündür. (Pnömotorakslar*: Akciğerdeki havanın çeşitli nedenlerle akciğer dışı dokulara kaçışı.)
Ehlers-Danlos Sendromdan en çok etkilenen yapılar
- Cilt: Ehlers-Danlos sendromundan etkilenen hastalarda cilt hyperelastic olarak bilinen boyun, yüz ve eklemlerdeki deri birkaç santimetreye kadar esnetilebilir ve serbest bırakıldığında geri çekilir. Ayrıca yaralar geç iyileşir ve yara kenarları genişleyerek atrofik alt yara izleri oluşur
- Eklemler: Eklemler normalin üzerinde hareket genişliğine sahiptir (hipermobilite). Eklemler aşırı genişleyerek tuhaf hareketler gerçekleştirebilir. Çıkıklar ve yanlış hizalamalar yaygındır.
- Damarlar: Hem küçük hem de büyük damarlar hamilelik, doğum veya spor esnasında olası bir travma nedeniyle ile kolayca yırtılarak tehlikeli kanamalara sebep olabilir.
Teşhis
- Doktor muayenesi
- Genetik inceleme
- Deri biyopsisi
- Görüntüleme prosedürleri
Ehlers-Danlos sendromunun teşhisi, fiziki muayeneden elde edilen semptom ve bulgulara dayanır. Bu bulgular genellikle birbirine benzer yada yakın bulgular olduğu için hastalığın hangi alt tipe ait olduğu genellikle genetik test ile teyit edilmesi gerekir.
Ehlers-Danlos sendromunun bazı formları cilt örnekleri mikroskop altında incelenerek deri biyopsisi yöntemi ile belirlenebilir.
Hastalığın seyri
Ehlers-Danlos sendromunun birçok farklı komplikasyonu olmasına rağmen hastaların çoğunda normal bir yaşam beklentisi vardır. Ancak belirli formlardaki komplikasyonlar nadir de olsa ölümcül olabilmektedir (genellikle kanama). Bazı formlarda ise hastanın yaşam kalitesi sınırlıdır.
Hamilelerde durum: Hamilelik öncesi doktor ile konuşularak hamileliğin ve doğumun bu konuda uzman bir kadın doğum uzmanı denetiminde gerçekleşmesi sağlanmalıdır. Ayrıca aile üyelerine genetik danışmanlık önerilir.
Ehlers-Danlos-Syndrom sınıflandırması
EDS sınıflandırma çalışmaları ilk olarak 1960’ların sonlarına doğru başladı. Ehlers-Danlos sendromu 1997 yılında Villefranche sınıflandırması adı verilen bir sınıflandırma ile alt bölümlere ayrıldı ve bu sınıflandırma 1988’de Berlin’de bir konferansta yayınlandı. Villefranche sınıflandırması ile hastalık ana ve ikincil kriterler olmak üzere 6 tip ile tanımlandı. Bu sınıflandırma hastalığın klinik teşhisini kolaylaştırarak benzer diğer hastalıklardan ayırt etmeye yardımcı oldu.[1]
2017 yılında Villefranche sınıflandırma tamamen revize edilerek yerini yeni tanı kriterleri aldı ve hastalık 13 alt tip ile tanımlandı. [2]
Yeni tanı kriterlerine göre artık Oksipital boynuz sendromu, Fibronektin eksikliği (EDS tıp X), ailesel Hipermobilite sendromu (EDS tip XI), X’e bağlı EDS (EDS tip V) ve Filamin A EDS Ehlers-Danlos sendromu dan sayılmıyor. [3]
Not: Bu alt tiplerin semptomlarında büyük değişiklik ve örtüşmeler bulunuyor. Bu nedenle net bir sınıflandırma, yalnızca klinik teşhis, genetik testler veya deri biyopsileri ile mümkündür.
Villefranche sınıflandırma 1997
Otozomal Dominant kalıtım yolu ile:
- Klasik tip, Gene COL5A1 ve COL5A2 genleri
- Hipermobil tip, gen bilinmiyor
- Vasküler tip, COL3A1 geni
- Artrokalazik tip, genler COL1A1 ve COL1A2 genleri
Otozomal resesif kalıtım ile:
- Kifoskolyotik tip, gen PLOD1
- Kermatosparaxic tip, ADAMTS2 geni
2017 deki uluslararası tanı kriterlerine göre sınıflandırma
1- Klasik EDS (cEDS) (eski adıyla tip 1 ve 2): Genetik nedenli EDS lerin yaklaşık % 90’ı COL5A2 ve COL5A genlerindeki mutasyon dan kaynaklanır. COL1A1 deki mutasyonlar nadirdir. Klasik EDS, otozomal dominant modelde kalıtılır. [4]
Yaygın semptomlar:
- Oldukça elastik ve kolayca çürükler oluşturan pürüzsüz, kadifemsi cilt
- Anormal yara iyileşmesi
- Sıklıkla çıkıklara (luksasyonlara) ve kısmi çıkıklara (subluksasyonlara) yol açan eklem hipermobilitesi
- Dirsek gibi basınç noktalarında kireçlenmiş hematomlar (Molluscoid pseudotumors)
- Genellikle ön kollarda ve / veya kaval kemiğinde görülen yağlı kistler (Deri altı sferoidler)
- Anormal derecede düşük kan basıncı (Hipotansiyon)
- Gecikmiş motor gelişimi
- Fıtıklara, rektal sarkmaya ve diğer komplikasyonlara yol açabilen doku kırılganlığı
- Kanı kalpten vücudun geri kalanına dağıtan kan damarının genişlemesi
- Fetal membranların doğum başlamadan önce yırtılarak hamileliğin zora girmesi (Erken membran rüptürü)
2- Klasik benzeri EDS (clEDS): TNXB genindeki mutasyonlardan kaynaklanır ve bu gen clEDS ile ilişkili tek gendir. Klasik benzeri EDS, otozomal resesif modelde kalıtılır. Kesin teşhis için genetik test gereklidir. [5]
Yaygın semptomlar:
- Eklemlerin aşırı esnekliği
- Yorgunluk
- Düşük veya zayıf kas tonusu (Hipotoni)
- Zayıf üst kol ve üst bacak kasları
- Kolay morarma
- Kas ağrıları
- Hiperelastik ve ince cilt
- İskelet kası dejenerasyonu
- Duyusal nöropati (duyu sinirlerinde hasar)
- Eklem ağrısı
- Aşırı gerilebilir ve kadifemsi bir cilt
- Genellikle omuz ve ayak bileklerinde tekrarlayan çıkıklar
- Cilt kolayca yaralanabilir veya kendiliğinden ekimozlar oluşur
3- Kardiyak-valvüler EDS (cvEDS): COL1A2 genindeki mutasyonlardan kaynaklanır ve bu gen cvEDS ile ilişkili tek gendir. Kardiyak-valvüler EDS, otozomal resesif modelde kalıtılır. Kesin teşhis için genetik test gereklidir.
Yaygın semptomlar:
- Kalp kapakçıklarının giderek zayıflaması
- Yüksek tansiyon veya arter hastalığı
- Kolayca moraran ince cilt
- Eklemlerin kararsızlığı, tekrarlayan çıkıklar veya kırıklar, yürüme zorluğu ve ellerde zayıf tutuş
- Skolyoz veya omurganın anormal yana doğru eğriliği
- Şiddetli ve ilerleyen kalp kapak sorunları (aort kapağı, mitral kapak)
- Deride aşırı uzayabilirlik
- Atrofik skarlar, ince deri, kolay morarma
- Eklem hipermobilitesi [5]
4- Vasküler EDS (vEDS) (eski adıyla tip 4): COL3A1 genindeki heterozigot bir mutasyon sebep olur. Vasküler EDS, otozomal dominant modelde kalıtılır.
Yaygın semptomlar:
- Arterin balon gibi genişlemesi(anevrizma), diseksiyon veya rüptür (damar iç duvar veya bir iç organın yırtılması)

- Bağırsak yırtılması
- Hamilelik sırasında uterus rüptürü(yırtılması
- İnce, yarı saydam hematoma belirgin eğilimli cilt (özellikle göğüs / karın bölgesinde fark edilir)
- Karakteristik yüz görünümü (dudaklarda ince vermilyon, mikrognati, dar burun, belirgin gözler)
- Ekstremitelerde, özellikle ellerde yaşlı görünüm (Acrogeria)
- Göz damarlarında anomaliler (Karotis-kavernöz sinüs arteriyovenöz fistül)
- Tendon / kas rüptürü
- Erken başlangıçlı varisli damarlar
- Akciğerleri çevreleyen plevral boşlukta kan ve hava birikimi (Pnömotoraks / hemopnömotoraks)
- Kolay morarma (spontan veya minimal travma ile)
- Kronik eklem çıkıkları (subluksasyonları)
- Kalçanın doğuştan çıkığı
- Çarpık ayak (Talipes ekinovarus)
- Küçük eklemlerde aşırı hareketlilik (hipermobilitesi) [5] [18]
5- Hipermobil EDS (hEDS) (eski adıyla tip 3): Hipermobil EDS’in genetik neden henüz bilinmemektedir. Hipermobil EDS, otozomal dominant modelde kalıtılır. Hipermobil EDS (hEDS) tanısı için henüz tanımlanmış gen bulunamamıştır o yüzden hEDS tanısı ancak klinik bulgularla yapılır.
Yaygın semptomlar:
- İpeksi bir cilt ve ciltte hafif esneme
- Hem büyük (dirsek ve dizler) hem de küçük (el ve ayak parmak) eklemleri etkileyen eklem hipermobilitesi
- Sıklıkla omuz, diz kapağı ve / veya alt çeneyi kafatasına bağlayan eklemde eklem çıkıkları ve subluksasyonları (kısmi çıkıklar)
- Hafif esnek ve kolayca moraran yumuşak, pürüzsüz cilt
- Kronik kas-iskelet ağrıları
- Erken başlayan kireçlenme (osteoartrit)
- Kemik erimesi (osteoporoz)
- Bulantı, şişkinlik, kusma, mide ekşimesi, kabızlık veya mide fıtığı gibi gastrointestinal sorunlar ayrıca mide ekşimesi veya reflü gibi sorunlar (Dismotilite)
- Otonom sinir sisteminde işlev bozukluğu
- Mitral kapak prolapsusu veya aort kökü genişlemesi gibi kardiyovasküler anormallikler (kanı kalpten vücudun geri kalanına dağıtan kan damarının genişlemesi)
- Kadınlarda pelvik prolapsus, ağrılı adet kanaması (dismenore) ve ağrılı ilişki (disparoni)
- Erken membran rüptürü veya hızlı doğum sancıları ve artan gebelik komplikasyonları
- Alt eklemlerde sıklıkla çıkıklar
- Kronik uzuv ve eklem ağrısı
- Tendon iltihabı [19] [20] [21] [22]
6- Arthrochalasia EDS (aEDS) (eski adıyla tip 7A ve 7B): COL1A1 veya COL1A2’deki heterozigot mutasyonlardan kaynaklanır. Bu mutasyon, ilgili genin 6. Ekzonu’nun tamamen veya kısmen kaybına neden olur. aEDS, bu iki gen dışında başka bir gen ile ilişkilendirilmemiştir. Kesin teşhis için genetik test gereklidir. aEDS otozomal dominant modelde kalıtılır.
Yaygın semptomlar:
- Konjenital bilateral kalça çıkığı
- Çoklu ve kısmi çıkıklar
- Ciltte aşırı uzayabilirlik [5]
7- Dermatosparaksis EDS (dEDS) (eski adıyla tip 7C): ADAMTS2 deki bialelik mutasyonlardan kaynaklanır ve ADAMTS2 geni dEDS ile ilişkili tek gendir. dEDS otozomal resesif modelde kalıtılır.
Yaygın semptomlar:
- Son derece kırılgan olan yumuşak, hamurlu cilt, ciltte şiddetli morarma ve yara izi, özellikle yüzde sarkık ve fazla deri
- Kısa boy, kısa parmaklar
- Kabarık göz kapakları, mavi skleralar (gözlerin beyazları)
- Bebeklerde kafatasının büyüyeceği alan olan fontanellerin/bıngıldak gecikmeli kapanması
- Aşağı doğru eğimli palpebral fissürler (gözlerin aşağıya bakan dış köşeleri)
- Alt çenenin yetersiz gelişmesi (Mikrognati) ile karakteristik yüz görünümü
- Mesane veya diyaframın yırtılması
- Hafif ila şiddetli eklem hipermobilitesi, fıtık [6] [7] [8]
8- Kifoskolyozik EDS (kEDS) (eski adıyla tip 6): PLOD1 geninde ve nadiren FKBP14 genindeki mutasyonlardan kaynaklanır. [5] [9] [23 ] kEDS, otozomal resesif modelde kalıtılır.
Yaygın semptomlar:
- Kırılgan ve kolayca moraran aşırı uzayabilir cilt.
- Sık luksasyon ve subluksasyonlara (çıkık ve kısmı çıkık) yol açan eklem hipermobilitesi
- Doğumda şiddetli hipotoni (kasın harekete karşı gösterdiği direnç)
- Doğumda veya yaşamın ilk yılında ortaya çıkan progresif kifoskolyoz (Omurganın göğüs veya bel bölgelerinde yana doğru eğilmesi)
- Skleral kırılganlık (göz küresine koruyucu kılıfın yırtılması)
- Yara iyileşmelerinde anormallikler
- Uzun, ince parmaklar (Marfanoid habitus), alışılmadık derecede uzun uzuvlar (Arachnodactyly) ve batık bir göğüs (pectus excavatum) veya çıkıntılı göğüs (pectus carinatum)
- Gecikmiş motor gelişimi
- Kırılmaya yatkın kırılgan arterler(damarlar) ve alışılmadık derecede küçük kornealar
- Mitral kapak prolapsusu (Kalbin kasılması sırasında kalp kapakçığının sol kulakçığa doğru bombeleşmesi, kubbeleşmesi ya da çökmesi)
- Aort kökü genişlemesi. Kardiyovasküler anormallikler (kanı kalpten vücudun geri kalanına dağıtan kan damarının genişlemesi)
- Düşük kemik yoğunluğu (Osteopeni)
- Doğuştan çarpık ayak
9- Kırılgan kornea sendromu (BCS): 2 tip BCS vardır. BCS tip 1, ZNF469 genindeki mutasyonlardan, BCS tip 2 ise PRDM5 genindeki mutasyonlardan kaynaklanır. BCS tanısı semptomlara göre yapılır ve genetik testlerle doğrulanır. BCS, otozomal resesif bir şekilde kalıtılır.
testlerle doğrulanır. BCS, otozomal resesif bir şekilde kalıtılır.
Yaygın semptomlar:
- Gözün koruyucu dış tabakasının (kornea) incelmesini ve buna bağlı olarak korneada küçük bir hasar ve ardından yırtılma
- Uzağı görememe (miyop), gözlerin beyaz kısmında mavimsi bir ton (mavi sklera) ve retina dekolmanı
- İşitme kaybı
- Kalça kemiklerinin anormal konumlanması (kalça displazisi)
- Anormal yara izi ve yumuşak cilt [10] [5]
10- Spondylodysplastic EDS (spEDS): B4GALT7, B3GALT6 veya SLC39A13’daki genlerindeki mutasyonlardan kaynaklanabilir. Ehlers-Danlos sendromları öncelikle cildi, saçlar ve iskelet sistemini etkiler. Belirtiler genellikle çocukluk veya ergenlik döneminde başlar
Yaygın semptomlar:
- Çocukluk ve yetişkinlikte kısa boy (152 cm den az)
- Zayıf kas tonusu (hipotoni)
- Uzuvların eğilmesi
- Yumuşak, ince, yarı saydam aşırı esnek bir cilt
- Pes planus (düz taban)
- Gecikmiş motor gelişimi
- Düşük kemik-mineral yoğunluğu nedeniyle kırılgan kemikler(Osteopeni)
- Hafif düzeyde zihinsel engel veya öğrenme güçlüğü
- Göz problemleri
- Karakteristik yüz özellikleri (üçgen yüz, geniş gözler, şişkin gözler ( proptozis), dar ağız, alçak kulaklar, kafada seyrek kıllar, düz yüz, geniş alın, mavi sklera, anormal dişler ve yarık damak (bifid uvula)
- İnce, kıvırcık saçlar, seyrek kaşlar ve kirpikler
- Eklem kontraktürleri ve hipermobilite
- Yüzde gevşek, elastik cilt [5] [11] [15]


11- Musculocontractural EDS (mcEDS): CHST14’teki mutasyonlardan kaynaklanır. Benzer bir fenotipe sahip hastalarda DSE geninde birkaç mutasyon tanımlanmıştır. Otozomal resesif bir bağ dokusu bozukluğudur. Kesin teşhis için genetik test gereklidir.[5] [16] [17]
Yaygın semptomlar:
- Adducted başparmak ve çarpık ayak sendromu (ATCS)
- Konjenital malformasyonlar, başparmak ve ayaklarda kontraktürler
- Tipik bir yüz görünümü ama normal bilişsel gelişim
- Kasların kısalması ile oluşan eklem deformasyonu (Multipl Konjenital Eklem Kontraktürleri) karakteristik olarak addüksiyon-fleksiyon kontraktürleri ve / veya talipes ekinovarus (çarpık ayak)
- Doğumda veya erken bebeklik döneminde belirgin kraniyofasiyal (kafatası ve yüz) özellikler
- Cildin aşırı uzayabilirliği, kolay morarma, atrofik izlerle cilt kırılganlığı, artmış palmar kırışıklığı gibi karakteristik kutanöz özellikler
12- Miyopathik EDS (mEDS): COL12A1 genindeki heterozigot veya bialelik mutasyonlardan kaynaklanır. Kesin teşhis için genetik test gereklidir. Miyopatik EDS, otozomal dominant veya otozomal resesif modelde kalıtılır.
Yaygın semptomlar: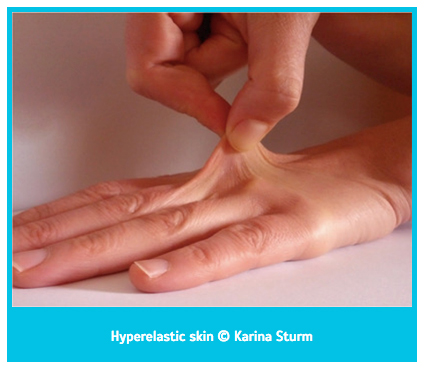
- Erken çocukluk döneminde diz, kalça ve dirsek eklemlerinde kas güçsüzlüğü
- Distal eklemler olarak bilinen ayak bilekleri, bilekler, ayaklar ve ellerin eklemlerinde hipermobilite
- Kas zayıflığı (yaşla birlikte gelişme eğiliminde)
- Yumuşak cilt ve ciltte girintili yar izleri (atrofik skarlar)
- Kas güçsüzlüğü, kramplar, sertlik ve spazmları içerebilen motor gelişim ve miyopati deki gecikmeler.
13- Periodontal EDS (pEDS): C1R veya C1S’deki heterozigot mutasyonlarından kaynaklanır. Periodontal EDS (pEDS), Ehlers-Danlos Sendromu nun en nadir formudur. Periodontal EDS (pEDS), otozomal dominant modelde kalıtılır. Kesin teşhis için genetik test gereklidir. [5]
Yaygın semptomlar:
- Erken başlangıçlı periodontitis (bu durum 30 yaşından önce diş kaybına neden olabilir)
- Kısa boy
- Ciltte oluşan koyu lekeler (hiperpigmente)
- Atrofik izler

- Aşırı uzayabilir cilt
- Aşırı esnek eklemler
- Dişlerden normalden daha küçük (mikrodonti)
- Diş etlerinin aşırı büyümesi
- Kalıcı dişlerin gelişmemesi

Tedavi
Ne nedensel, ne de semptomları ortadan kaldırmaya yönelik bir tedavi şimdilik mümkün değil. Etkilenen hastalar eklemlerde aşırı stresten kaçınmalı ve yavaş iyileşen yaralar nedeniyle yara almamaya dikkat etmelidirler .
Yaralanmayı önleyici belirli önlemler alınmalıdır:
- Şiddetli Ehlers-Danlos sendromu olan çocuklara koruyucu giysiler ve pedler giyebilirler.
- Ameliyat sadece acil durumlarda yapılmalıdır ve operasyon sırasında aşırı kanamayı karşı özel teknikler kullanılmalıdır.
- Hamilelik öncesi doktor ile konuşulmalı. Hamilelik ve doğum bir kadın doğum uzmanı hamileliği ve doğumu denetlemelidir.
Benzer konularda hazırlanmış diğer makaleler
- Genetik Hastalıklar: Charcot-Marie-Tooth (CMT)
- Genetik hastlıklar: Hemofili
- Genetik hastalıklar: Huntington
- Sigara alışkanlığı ve genetik yatkınlık
- Kromozomal hastalıklar-1 (Trizomi 21, Trizomi 18 ve Trizomi 13)
- Kromozomal Hastalıklar–2 (Angelman Sendromu, Prader-Willi Sendromu)
- Multipl skleroz(MS) nedir? Tanı ve tedavisindeki yeni bilimsel gelişmeler!
- Kırmızı şarap ve siyah üzüm, Multiple Skleroz(MS) hastalarının durumunu kötüleştiriyor.
- Alkolün kas gelişimine olumsuz etkisi
- Multiple Sklerose (MS) ile savaşta vitamin D
- Sporun Alzheimer’a karşı koruyucu etkisi
- Kokuların anıları çağrıştırması ve koku ile Alzheimer arasında ilişki
- Köpek balığı, Alzheimer ve Parkinson tedavisinde bir umut olabilir
- Alzheimer’a karşı alınacak yedi basit önlem
- Epilepsi hastaları için yeni bir umut : Gen terapisi
- Parkinson tedavisinde kullanılan bir ilaç, Multipl Skleroz tedavisinde de başarılı sonuçlar verdi
Mehmet Saltuerk
++++++++++++++++++++++++
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics
of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
- Oral health in prevalent types of Ehlers–Danlos syndromes
- 2017 eds international classification
- Eds types
- Ehlers-Danlos syndrome, classical type
- The Ehlers–Danlos Syndromes, Rare Types
- Clinical manifestations and diagnosis of Ehlers-Danlos syndromes
- Dermatosparaxis (Ehlers–Danlos Type VIIC): Prenatal Diagnosis Following a Previous Pregnancy With Unexpected Skull Fractures at Delivery
- The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC)
- Ehlers-Danlos Syndrome, Kyphoscoliotic Form.
- Brittle cornea syndrome: recognition, molecular diagnosis and management
- A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type
- Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid.
- A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome.
- Ehlers-Danlos features with progeroid facies and mild mental retardation: further delineation of the syndrome.
- The 2017 international classification of the Ehlers–Danlos syndromes.
- Loss of Dermatan-4-Sulfotransferase 1 Function Results in Adducted Thumb-Clubfoot Syndrome
- Congenital Disorders of Glycosylation with Emphasis on loss of Dermatan-4-Sulfotransferase
- Vascular Ehlers-Danlos Syndrome
- Hypermobile Ehlers-Danlos Syndrome
- Clinical manifestations and diagnosis of Ehlers-Danlos syndromes
- Ehlers Danlos syndrome and gastrointestinal manifestations: a 20-year experience at Mayo Clinic
- Gastrointestinal problems in hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders
- Ehlers Danlos syndrome, kyphoscoliotic type due to Lysyl Hydroxylase 1 deficiency in two children without congenital or early onset kyphoscoliosis
Bu blogdaki makaleler bir başka yayın organında kaynak gösterilmeden yayınlanamaz, çoğaltılamaz ve kullanılamaz.

Merhaba Mehmet bey oğlumun dirsekleri hypermobile uzun boy kollar marfanoid habitus mvp skolyoz sbo şüpheli pseudotumor var genetik de önce Col12a1 mutasyon sonra yapılan wes de Col12a1 mutasyon yok denildi. Sanger le doğrulama yapmadılar wes de çıkmadığı için. Meds olmasa bile heds olma ihtimali yüksek ama türkiyede klinik olarak bu teşhisi koyabilecek doktor yok bana ne tavsiyee edersiniz dataları başka bir laba tekrar analiz? Yada tüm genlerin büyük deletion ve do ip icasyonları için gerekli test? Yada bağdokusu mpaneli 60 geni içeren? Yada genom analizi? Yada trio anne baba çocuk. Ben en azından genetik olarak tespit edilebilen bir bağ dokusu hastalığı olmadığından emin olmak istiyorum.
Sayın Nihan Bağça, belirtier bağ dokusu ile ilgili bir problem olduğunu düşündürüyor. Bu konudan bir çok gen Sorumlu.
Sanırım size labordan tek sayfalık bir rapor verilmiştir. 0ysa yapılan Wes analızine birçok gen ile ilgili Sonuçlar vardır. Bunlara dikkatlice balmak gerek. Yani Wes analızlerinin ham datalarına bir balmak gerek. Belki orda bir şeyler bulunabilir.